New GMP Cleanroom Classification &Supervision: The Complete FAQ Guide in 2025
According to the Good Manufacturing Practice (GMP) for pharmaceutical production, the air cleanliness level in the GMP cleanroom is determined by the production process. The production environment parameters in the cleanroom for pharmaceutical production, such as temperature, relative humidity, and pressure difference, are generally determined by the production process. Generally, the temperature is 18℃~24℃, and the relative humidity is 45%~65%.
The implementation guidelines of GMP are relatively specific. The temperature and relative humidity in a clean pharmaceutical production plant are based on the principle that operators wearing clean work clothes do not feel uncomfortable.
Well, let’s explore the new GMP cleanroom classification & supervision together!
1.The Four Level of Cleanroom

Cleanroom - Sourced: starrco
A-level: High-risk operation areas, such as filling areas, areas where rubber stoppers are placed and open packaging containers that come into direct contact with sterile preparations, and areas where sterile assembly or connection operations are carried out, should be maintained in a one-way flow operating platform (cover) to maintain the environmental state of the area. The one-way flow system must provide uniform air supply in its working area, with a wind speed of 0.36-0.54m/s (guidance value). There should be data to prove the state of one-way flow and verified. In a closed isolation operator or glove box, you can use lower wind speeds.
B-level: it refers to the background area where A-level areas are located for high-risk operations such as aseptic preparation and filling.
C-level and D-level: it refers to a clean area in the production process of sterile drugs where the importance of the operation steps is relatively low.
The standard regulations for air airborne particle of the above level are shown in the table below:
| Cleanliness Level | Maximum allowable number of airborne particle per cubic meter | |||
| Statics | Dynamics | |||
| ≥0.5 μm | ≥5.0 μm | ≥0.5 μm | ≥5.0 μm | |
| A-level (1) | 3,520 | 20 | 3,520 | 20 |
| B-level | 3,520 | 29 | 352,000 | 2,900 |
| C-level | 352,000 | 2,900 | 3,520,000 | 29,000 |
| D-level | 3,520,000 | 29,000 | Not specified | Not specified |
(1) To confirm the classificationof A-level clean area, the sampling amount at each sampling point shall not be less than 1 cubic meter. The classification of airborne particle in A-level clean areas is ISO 4.8, with airborne particle ≥ 5.0 μm as the limit standard. The classification in B-level clean areas (static) is ISO 5, which includes airborne particle of two particle sizes as shown in the table. For C-level clean areas (static and dynamic), the classifications are ISO 7 and ISO 8, respectively. For D-level clean areas (static), the classification is ISO 8. The testing method can refer to ISO14644-1.
(2) When confirming the classification, you should use a portable dust particle counter with a shorter sampling tube to prevent airborne particleof ≥ 5.0 μm from settling in the long sampling tube of the remote sampling system.
(3) Dynamic testing can be conducted during routine operations and simulated filling of culture media to demonstrate the achievement of dynamic cleanliness levels. However, simulated filling of culture media requires dynamic testing to be conducted under the “worst” condition.
Airborne Particle - Sourced: LUMIAIR Singapore
Article 10: Dynamic monitoring of airborne particle in clean areas shall be carried out in accordance with the following requirements:
(1) Based on the results of cleanliness level and air purification systemand risk assessment, determining the location of sampling points and conduct daily dynamic monitoring.
(2) Throughout the entire critical operation process, including equipment assembly, suspended particle monitoring should be carried out in the A-level clean area. When pollution during the production process (such as live organisms and radiation hazards) may damage the dust particle counter, testing should be conducted during equipment debugging and simulation operations.
(3) A monitoring system similar to that of an A-level clean area can be used in B-level clean areas. The sampling frequency and amount can be adjusted based on the degree of impact of B-level clean areas on adjacent A-level clean areas.
(4) The influence of the length of the sampling tube and the radius of the bend on the test resultsshould be considered in the monitoring system for airborne particle.
(5) The sampling amount for daily monitoring can be different from the air sampling amount for cleanliness level and air purification system.
(6) When a small amount of airborne particle≥ 5.0 µm appear continuously or regularly in A-level and B-level clean areas, an investigation should be conducted.
(7) After all production operations have been completed, the operators have withdrawn from the production site and self cleaned for 15-20 minutes (guidance value), the airborne particlein the clean area should meet the “static” standard in the table.
(8) Dynamic monitoring of C-level clean areas and D-level clean areas (if necessary) should be carried out in accordance with the principles of quality risk management. The monitoring requirements, warning limits, and correction limits can be determined based on the nature of the operation, but the self purification time should meet the specified requirements.
(9) Temperature, relative humidity, and other parameters should be formulated based on the nature of the product and operation, and these parameters should not have a negative impact on the specified cleanliness.

Microbial Monitoring - Sourced: pharmaceuticalonline
Article 11: Dynamic monitoring of microorganisms shall be conducted to evaluate the microbial status of sterile production.
The monitoring methods include settling bacteria method, quantitative air airborne microbe sampling method, and surface sampling method (such as cotton swab wiping method and contact plate method). Dynamic sampling should avoid adverse effects on clean areas.
The review of finished product batch records should include the results of environmental monitoring. The monitoring of the surface and operators should be carried out after the completion of critical operations. In addition to normal production operation monitoring, microbial monitoring can be added after system validation, cleaning, or disinfection operations are completed.
The dynamic standards for microbial monitoring in clean areas are as follows:
| Cleanliness Level | Airborne Microbe
cfu/m3 |
Settling Microbe(f90mm)
cfu /4小时(2) |
Surface Microorganisms | |
| Contact(f55mm)
cfu /plate |
5-Finger Gloves cfu /Gloves | |||
| A-level | <1 | <1 | <1 | <1 |
| B-level | 10 | 5 | 5 | 5 |
| C-level | 100 | 50 | 25 | - |
| D-level | 200 | 100 | 50 | - |
Note: (1) All values in the table are averages.
(2) The exposure time of a single settling plate can be less than 4 hours, and multiple settling plates can be used for continuous monitoring and cumulative counting at the same location.
Article 12: Appropriate warning and correction limits for monitoring airborne particle and microorganisms shall be established. The operating procedures should specify in detail the corrective measures to be taken when the results exceed the standard.
Article 13: The production and operation environment of sterile drugs can be selected according to the examples in the table.
| Cleanliness Level | Example of production operation for final sterilized products |
| Local A-level in C-level background | Filling (or sealing) of products with high pollution risk (1) |
| C-level | 1. Product filling (or sealing);
2. High pollution risk (2) product formulation and filtration; 3. Preparation and filling (or sealing) of eye preparations, sterile ointments, sterile suspensions, etc; 4. Handling of packaging materials and utensils that come into direct contact with drugs after final cleaning. |
| D-level | 1. Rolling cap;
2. Preparation of materials before filling; 3. The final cleaning of packaging materials and utensils that come into direct contact with drugs through product preparation (referring to concentrated preparation or preparation using a closed system) and filtration. |
Note: (1) The high pollution risk here refers to situations where the product is prone to bacterial growth, filling speed is slow, the filling container is a wide mouthed bottle, and the container must be exposed for several seconds before being sealed;
(2) The high pollution risk here refers to situations where the product is prone to bacterial growth, requires a long wait after preparation before sterilization, or is not prepared in a closed system.
| Cleanliness Level | Examples of sterile production operations for non final sterilized products |
| A-level under B-level background | 1. The operation and transportation of products in an incompletely sealed (1) state, such as product filling (or sealing), packaging, plugging, capping (2), etc;
2. Preparation of drugs or products that cannot be sterilized or filtered before filling; 3. Packaging materials that come into direct contact with drugs, assembly of sterilized instruments, and transportation and storage in an incompletely sealed state; 4. Crushing, sieving, mixing, and packaging of sterile raw materials. |
| B-level | 1. Transfer of products that are not fully sealed (1) in a fully sealed container;
2. Transfer and storage of packaging materials and instruments that come into direct contact with drugs in sealed containers after sterilization. |
| C-level | 1. Preparation of fungicides or products that can be sterilized and filtered before filling;
2. Filtering of products. |
| D-level | The final cleaning, assembly, packaging, and sterilization of packaging materials and equipment that come into direct contact with drugs. |
Note: (1) Before capping, the product is considered to be in an incomplete sealed state.
(2) Based on factors such as the sealing performance of the already compressed product, the design of the capping equipment, and the characteristics of the aluminum cap, the capping operation can be carried out in an A-level air supply environment with a C-level or D-level background. The A-level air supply environment should at least meet the static requirements of the A-level area.
2.Isolation Operation Technology

Isolation operation technology - Sourced: telstar
Article 14: High pollution risk operations should be completed in isolation operators. The design of the isolation operator and its environment should ensure that the air quality in the corresponding area meets the set standards. The transmission device can be designed as a single or double door, or as a fully sealed system connected to the sterilization equipment.
Special attention should be paid to preventing contamination during the entry and exit isolation of items.
The environment in which the isolation operator is located depends on its design and application. The environment in which the isolation operator for sterile production is located should be at least a D-level clean area.
Article 15: Isolation operators can only be put into use after appropriate confirmation. When confirming, all key factors of isolation technology should be considered, such as the air quality of the environment inside and outside the isolation system, disinfection of isolation operators, transfer operations, and the integrity of the isolation system.
Article 16: Isolation operators and isolation sleeve or glove systems shall undergo routine monitoring, including frequent necessary leak testing.
3.Blow-Fill-Seal (BFS) Technology
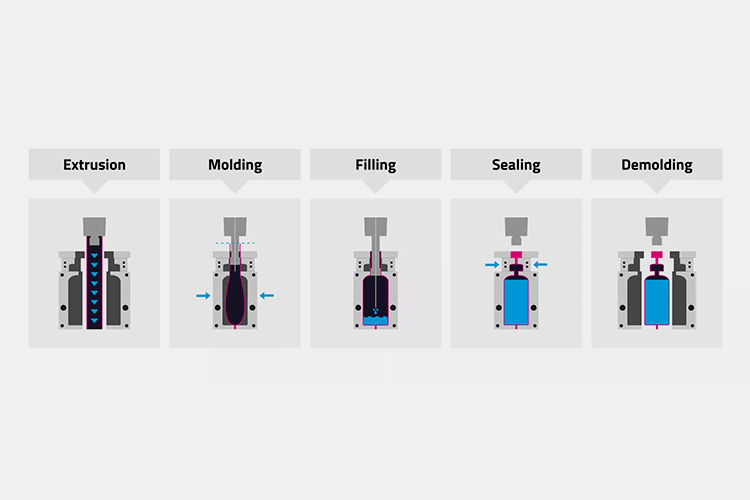
Blow-Fill-Seal (BFS) Technology - Sourced: pharmsolution
Article 17: Blow-Fill-Seal equipment used for the production of non final sterilized products should be equipped with A-level air shower devices, and personnel attire should conform to the style of A/B-level clean areas. The equipment should be installed at least in a C-level clean area environment. Under static conditions, both airborne particle and microorganisms in this environment should meet the standards, while under dynamic conditions, microorganisms in this environment should meet the standards. The BFS equipment used for producing final sterilized products should be installed at least in a D-level clean area environment.
Article 18: Due to the special nature of BFS technology, special attention should be paid to the design and confirmation of equipment, verification and reproducibility of online cleaning and sterilization, clean area environment of equipment, training and dressing of operators, as well as operations in key areas of equipment, including sterile assembly of equipment before filling begins.
4.Personnel

Personal - Sourced: cleanroom-industries
Article 19: The number of people in the clean area shall be strictly controlled, and inspections and supervision shall be conducted as much as possible outside the sterile production clean area.
Article 20: Personnel working in clean areas (including cleaners and equipment maintenance workers) shall receive regular training to ensure that the operation of sterile drugs meets the requirements. The training content should include basic knowledge in hygiene and microbiology. Untrained external personnel (such as external construction or maintenance personnel) should be given special and detailed guidance and supervision when entering the clean area during production.
Article 21: Personnel engaged in animal tissue processing or personnel engaged in microbial cultivation unrelated to current production are generally not allowed to enter the sterile drug production area. When unavoidable, relevant personnel purification operating procedures should be strictly followed.
Article 22: Employees engaged in the production of sterile drugs shall report any abnormal situations that may cause pollution at any time, including the type and degree of pollution. When an employee’s health condition may increase the risk of microbial contamination, appropriate measures should be taken by designated personnel.

Work Clothes - Sourced: cphi-online
Article 23: Changing clothes and washing hands shall be carried out in accordance with the operating procedures, and pollution to the clean area shall be minimized or pollutants shall be brought into the clean area as much as possible.
Article 24: Work clothes and their quality shall be suitable for the requirements of production operations and the cleanliness level of the operating area, and their style and wearing method shall be able to meet the requirements of protecting products and personnel. The dress code requirements for each clean area are as follows:
| D-level clean area | Relevant parts such as hair and beard should be covered. Suitable work clothes and shoes or shoe covers should be worn. Appropriate measures should be taken to avoid pollutants brought into the clean area. |
| C-level clean area | Relevant parts such as hair and beard should be covered, and masks should be worn. You should wear a tight fitting jumpsuit or separate work clothes at the wrist, and wear appropriate shoes or shoe covers. Work clothes should not shed fibers or particles. |
| A/B level clean area | Relevant parts such as hair and beard should be covered with a hood, which should be tucked into the collar. A mask should be worn to prevent the emission of droplets, and protective goggles should be worn if necessary. Rubber or plastic gloves that have been sterilized and free of particles (such as talcum powder) should be worn, and sterilized or disinfected foot covers should be worn. |
Article 25: Personal outerwear shall not be brought into the changing rooms leading to B-level or C-level clean areas. Each employee should change their sterile work clothes every time they enter the A/B level clean area; Or at least once per shift, but the feasibility of this method should be demonstrated through monitoring results. During operation, gloves should be disinfected regularly and masks and gloves should be replaced if necessary.
Article 26: The cleaning and handling methods of work clothes used in clean areas shall ensure that they do not carry pollutants and do not pollute the clean area. The cleaning and sterilization of work clothes should be carried out in accordance with relevant operating procedures, and it is best to set up a separate laundry room.
5.Workshop

Workshop - Sourced: spectral.blue
Article 27: The design of a clean workshop should avoid unnecessary entry of management or monitoring personnel as much as possible. The design of B-level clean areas should enable management or monitoring personnel to observe internal operations from the outside.
Article 28: In order to reduce dust accumulation and facilitate cleaning, there shall be no difficult to clean parts such as shelves, cabinets, and equipment in the clean area. The design of the door should be easy to clean.
Article 29: It is prohibited to set up water tanks and floor drains in A/B level clean areas for aseptic production. In other clean areas, water tanks or floor drains should have appropriate design, layout, and maintenance, and should be equipped with easy to clean and air blocking devices to prevent backflow. The connection method with the external drainage system should be able to prevent the invasion of microorganisms.

Clean Workshop - Sourced: cleanroom-industries
Article 30: The changing room shall be designed in an air lock manner to separate the different stages of changing, and to avoid contamination of work clothes by microorganisms and particles as much as possible. The changing room should have sufficient ventilation times. The static level of the rear section of the changing room should be the same as the level of its corresponding clean area. If necessary, separate the changing rooms for entering and leaving the clean area. In general, hand washing facilities can only be installed in the first stage of changing clothes.
Article 31: The doors on both sides of the air lock room shall not be opened simultaneously. Chain systems or optical or (and) acoustic alarm systems can be used to prevent the doors on both sides from opening simultaneously.
Article 32: In any operating state, the clean area should be able to ensure positive pressure to the surrounding low-level areas through appropriate air supply, maintain good airflow direction, and ensure effective purification capacity.
Special protection should be given to the cleaned packaging materials and utensils that come into direct contact with the product, as well as the operating areas where the product is directly exposed.

Cleanroom - Sourced: pcne.eu
Article 33: It should be able to prove that the air flow method used does not cause pollution risks and there should be records (such as videos of smoke and fog tests).
Article 34: An alarm system for the failure of the air supply unit should be installed. Differential pressure gauges should be installed between adjacent grade zones where differential pressure is crucial. Differential pressure data should be regularly recorded or included in relevant documents.
Article 35: If a large amount of particles are generated during capping, a separate capping area should be set up and appropriate ventilation devices should be installed. If the capping area is not set separately, it should be able to prove that the capping operation has no adverse impact on product quality.
6.Equipment

Aipak Engineering BFS Equipment
Article 36: Except for the conveyor belt which can continuously sterilize (such as tunnel sterilization equipment), the conveyor belt shall not cross between A/B level clean areas and low-level clean areas.
Article 37: The design and installation of production equipment and auxiliary devices shall be as convenient as possible for operation, maintenance, and repair outside the clean area. Equipment that needs to be sterilized should be sterilized as much as possible after complete assembly.
Article 38: The air purification system in the clean area of sterile drug production shall maintain continuous operation and maintain the corresponding cleanliness level. If the air purification system is shut down and restarted due to reasons, necessary tests should be conducted to confirm that it still meets the specified cleanliness level requirements.
Article 39: When equipment maintenance is carried out in a clean area, if the cleanliness or sterility is damaged, cleaning, disinfection or sterilization should be carried out in the area. Production operations can only resume after the monitoring is qualified.
Article 40: Key equipment, such as sterilization cabinets, air purification systems, and process water systems, shall be confirmed and undergo planned maintenance before use.
Article 41: Filters should try their best not to shed fibers. It is strictly prohibited to use filters containing asbestos. The filter shall not have adverse effects on product quality due to reaction, release of substances or adsorption with the product.
Article 42: Production gases entering sterile production areas (such as compressed air and nitrogen, but excluding flammable gases) shall undergo sterilization and filtration, and the integrity of sterilization filters and respiratory filters shall be regularly checked.
7.Disinfect

Disinfect - Sourced: henderson-biomedical
Article 43: Clean areas shall be cleaned and disinfected in accordance with operating procedures. In general, there should be more than one type of disinfectant used. UV disinfection should not be used as a substitute for chemical disinfection. Regular environmental monitoring should be carried out to promptly identify tolerant strains and pollution situations.
Article 44: The microbial contamination status of disinfectants and cleaning agents shall be monitored, and the prepared disinfectants and cleaning agents shall be stored in clean containers for a period not exceeding the prescribed time limit. A/B grade clean areas should use sterile or sterilized disinfectants and cleaning agents.
Article 45: When necessary, fumigation may be used to reduce microbial contamination in sanitary blind spots in clean areas, and the residual level of fumigants shall be verified.
8.Production Management

Production Management - Sourced: mrpeasy
Article 46: Measures shall be taken at each stage of production (including the stages before sterilization) to reduce pollution.
Article 47: The validation of sterile production processes shall include simulated filling tests of culture media.
The selection of culture medium should be based on the dosage form of the product, the selectivity, clarity, concentration, and suitability for sterilization. It should simulate conventional aseptic production processes as much as possible, including all key operations that affect aseptic results, as well as various interventions and worst-case conditions that may occur during production.
The first validation of the simulated filling test of the culture medium should be conducted three consecutive qualified tests per shift. After significant changes in the air purification system, equipment, production process, and personnel, the simulated filling test of the culture medium should be repeated. The simulation filling test of the culture medium should usually be conducted once every six months according to the production process, with at least one batch per shift.

Culture Medium - Sourced: nucleusbiologics
The number of culture medium filling containers should be sufficient to ensure the effectiveness of the evaluation. For products with smaller batches, the quantity of culture medium filled should be at least equal to the batch size of the product. The goal of the culture medium simulation filling experiment is zero pollution, and the following requirements should be followed:
(1) When the filling quantity is less than 5,000 pieces, no contaminated products shall be detected.
(2) When the filling quantity is between 5,000 and 10,000 pieces:
- There is 1 piece of contamination that needs to be investigated, and repeated experiments can be considered;
- There are 2 contaminated samples that need to be investigated before further verification.
(3) When the filling quantity exceeds 10,000 pieces:
- There is one contamination that needs to be investigated;
- There are 2 contaminated samples that need to be investigated before further verification.
(4) Any microbial contamination should be investigated.
Article 48: Measures shall be taken to ensure that verification does not have adverse effects on production.
Article 49: The water used for the refinement of sterile raw materials, preparation of sterile drugs, final cleaning of packaging materials and instruments in direct contact with drugs, and preparation of disinfectants and cleaning agents in A/B level clean areas shall meet the quality standards for injection water.
Article 50: When necessary, regular monitoring of bacterial endotoxins in pharmaceutical water should be carried out, and relevant records of monitoring results and corrective measures taken should be kept.
Article 51: When aseptic production is in progress, special attention should be paid to reducing various activities in the clean area. Personnel movement should be reduced to avoid excessive particle and microbial emissions from vigorous activities. Due to the characteristics of the work clothes worn, the temperature and humidity of the environment should ensure the comfort of the operators.
Article 52: The degree of microbial contamination of materials shall be minimized as much as possible. When necessary, the quality standards of materials should include microbial limits, bacterial endotoxins, or pyrogen testing items.
Article 53: Containers and materials that are prone to fiber shedding should be avoided in clean areas; In the process of aseptic production, such containers and materials shall not be used.
Article 54: Various measures shall be taken to reduce particulate pollution in the final product.
Article 55: The handling of packaging materials, containers, and equipment after final cleaning shall avoid contamination again.
Article 56: The interval between cleaning, drying, and sterilization of packaging materials, containers, and equipment, as well as the interval from sterilization to use, shall be shortened as much as possible. Standard interval time control should be established under specified storage conditions.
Article 57: The interval between the preparation and sterilization (or sterilization filtration) of the drug solution should be shortened as much as possible. Corresponding interval control standards should be established based on the characteristics and storage conditions of the product.
Article 58: The monitoring standards for microbial contamination levels of products before sterilization shall be determined based on the effectiveness of the sterilization methods used, and regular monitoring shall be conducted. If necessary, pyrogens or bacterial endotoxins should also be monitored.
Article 59: Packaging materials, containers, equipment, and any other items used in aseptic production shall be sterilized and enter the aseptic production area through a double door sterilization cabinet or by other means, but contamination shall be avoided.

Cleanroom Faculty - Sourced: SORN
Article 60: Unless otherwise specified, the principles for batch division of sterile drugs are as follows:
(1) A batch of homogeneous products produced from the final preparation of the same solution tank for large (small) volume injections; If the same batch of products are sterilized using different sterilization equipment or the same sterilization equipment is sterilized in batches, they should be traceable;
(2) Powder injection is a batch of homogeneous products produced by a batch of sterile raw materials within the same continuous production cycle;
(3) Freeze dried products are a batch of homogeneous products produced using the same freeze-drying equipment in the same production cycle, using the same batch of prepared pharmaceutical solutions;
(4) Eye preparations, ointments, emulsions, suspensions, and other homogeneous products produced in the same preparation tank are considered as a batch.
9.Sterilization Process

Sterilization Process - Sourced: alabamabioclean
Article 61: Aseptic drugs shall be sterilized by heating as much as possible, and the probability of microbial survival (i.e. sterility assurance level, SAL) in the final sterilized product shall not exceed 10-6. For final sterilization using the moist heat sterilization method, the standard sterilization time F0 value should generally be greater than 8 minutes, and steam circulation treatment is not considered final sterilization.
For products with thermal instability, sterile production operations or alternative methods of filtration and sterilization can be used.
Article 62: Sterilization may be carried out using moist heat, dry heat, ion radiation, ethylene oxide, or filtration sterilization methods. Each sterilization method has its specific scope of application, and the sterilization process must be consistent with the requirements of registration and approval, and should be validated.
Article 63: Before any sterilization process is put into use, physical testing methods and biological indicators must be used to verify its applicability to the product or item and to verify that all parts have achieved sterilization effects.
Article 64: The effectiveness of sterilization processes shall be regularly revalidated (at least once a year). After major equipment changes, re verification is required. Re validation records should be kept.
Article 65: All items to be sterilized must be processed according to the prescribed requirements to achieve good sterilization effects, and the design of sterilization processes should ensure compliance with sterilization requirements.
Article 66: The loading method of products and items to be sterilized in the sterilization equipment chamber shall be confirmed through verification.
Article 67: Biological indicators shall be stored and used in accordance with the requirements of the supplier, and their quality shall be confirmed through positive control tests. When using biological indicators, strict management measures should be taken to prevent microbial contamination caused by this.
Article 68: There should be a clear method to distinguish between sterilized products and products to be sterilized. Each vehicle (pallet or other loading equipment) product or material should be labeled, clearly indicating the product name, batch number, and indicating whether it has been sterilized. If necessary, a damp heat sterilization indicator tape can be used to distinguish it.
Article 69: Each sterilization operation shall have a sterilization record and serve as one of the basis for product release.
10.Sterilization Method

Sterilization Method - Sourced: acmeresearchlabs
Article 70: Thermal sterilization usually includes moist heat sterilization and dry heat sterilization, which should meet the following requirements:
(1) During the verification and production process, temperature probes used for monitoring or recording and temperature probes used for control should be set separately, and the position of the set should be determined through verification.
(2) Chemical or biological indicators can be used to monitor sterilization processes, but they cannot replace physical testing.
(3) The heating time required for each loading method should be monitored, and the sterilization time should be calculated from the moment all sterilized products or items reach the set sterilization temperature.
(4) Measures should be taken to prevent contamination of sterilized products or items during the cooling process. Unless it can be proven that any leaking products or items can be removed during the production process, any cooling medium (liquid or gas) in contact with the products or items should be sterilized or sterilized.
Article 71: Moist heat sterilization shall meet the following requirements:
(1) The parameters for monitoring the moist heatsterilization process should include sterilization time, temperature, or pressure.
The sterilization cabinet with a drainage outlet at the bottom of the chamber should measure and record the temperature data of this point throughout the entire sterilization process if necessary. If vacuum operation is included in the sterilization process, regular leak testing should be conducted on the chamber.
(2) Except for sealed products, sterilized items should be appropriately wrapped with appropriate materials, and the materials and wrapping methods used should be conducive to air discharge, steam penetration, and prevent contamination after sterilization.
Article 72: Dry heat sterilization shall meet the following requirements:
(1) During dry heat sterilization, the air inside the sterilization cabinet chamber should circulate and maintain positive pressure to prevent non sterile air from entering. The air entering the chamber should be filtered through a high-efficiency filter, which should undergo integrity testing.
(2) When dry heat sterilization is used to remove pyrogens, validation should include a bacterial endotoxin challenge test.
(3) The temperature, time, and pressure difference inside and outside the chamber during the dry heat sterilization process should be recorded.

Radiation Sterilization - Sourced: alabamabioclean
Article 73: Radiation sterilization shall meet the following requirements:
(1) Radiation sterilization can only be used if it has been proven to have no adverse impact on product quality. Radiation sterilization should comply with the relevant requirements of the Pharmacopoeia of the People’s Republic of China and registration approval.
(2) The radiation sterilization process should be validated. The verification plan should include radiation dose, radiation time, packaging material, loading method, and examine the impact of packaging density changes on sterilization effectiveness.
(3) During the radiation sterilization process, a dose indicator should be used to measure the radiation dose.
(4) Biological indicators can serve as an additional monitoring tool.
(5) Measures should be taken to prevent confusion between irradiated and non irradiated items. There should be radiation indicator plates on each packaging that can produce color changes after radiation.
(6) The total radiation dose standard should be reached within the specified time.
(7) Radiation sterilization should be recorded.

Ethylene Oxide Sterilization - Sourced: gmdgroup
Article 74: Ethylene oxide sterilization shall meet the following requirements:
(1) Ethylene oxide sterilization should comply with the relevant requirements of the Pharmacopoeia of the People’s Republic of China and registration approval.
(2) The sterilization process validation should be able to prove that ethylene oxide does not cause destructive effects on the product, and the exhaust conditions and times set for different products or materials can ensure that all residual gases and reaction products are reduced to the set qualified limits.
(3) Measures should be taken to prevent microorganisms from being trapped in crystals or dry proteins, ensuring direct contact between sterilization gas and microorganisms.
(4) After the sterilized item reaches the temperature and humidity conditions specified in the sterilization process, sterilization gas should be introduced as soon as possible to ensure the sterilization effect.
(5) During each sterilization, appropriate and a certain amount of biological indicators should be placed in different parts of the sterilized item to monitor the sterilization effect. The monitoring results should be included in the corresponding batch records.
(6) The content of each sterilization record should include the completion time of the entire sterilization process, the pressure, temperature and humidity of the chamber during the sterilization process, the concentration of ethylene oxide, and the total consumption. The pressure and temperature of the entire sterilization process should be recorded, and the sterilization curve should be included in the corresponding batch record.
(7) The sterilized items should be stored in a controlled and ventilated environment to reduce residual gases and reaction products to the specified limits.

Requirements - Sourced: Smartsheet
Article 75: The filtration and sterilization of non final sterilized products shall meet the following requirements:
(1) Products that can be ultimately sterilized shall not replace the final sterilization process with filtration sterilization process. If the drug cannot be sterilized in its final packaging container, a 0.22 μm (smaller or equivalent filtration efficiency) sterilization filter can be used to filter the drug solution into a pre sterilized container. Due to the inability of sterilization filters to completely remove viruses or mycoplasma, heat treatment methods can be used to compensate for the shortcomings of sterilization filtration.
(2) Measures should be taken to reduce the risk of filtration and sterilization. It is advisable to install a second sterilized sterilization filter to filter the solution again, and the final sterilization filter should be as close to the filling point as possible.
(3) After using the sterilization filter, it is necessary to immediately check its integrity and record it using appropriate methods. The commonly used methods include bubble point test, diffusion flow test, or pressure retention test.
(4) The filtration and sterilization process should be validated, and the time required to filter a certain amount of drug solution and the pressure on both sides of the filter should be determined during the validation. Any significant deviation from normal time or pressure should be recorded and investigated, and the investigation results should be included in batch records.
(5) The time limit for the use of sterilization filters of the same specification and model should be verified and generally should not exceed one working day.
11.Final Treatment of Sterile Drugs
Final Treatment of Sterile Drugs - Sourced: Linkedin
Article 76: After the vial is corked, the capping should be completed as soon as possible. If leaving the sterile operation area or room before capping, appropriate measures should be taken to prevent product contamination.
Article 77: The sealing of sterile drug packaging containers shall be verified to avoid contamination of the product.
Sealed products (such as glass ampoule or plastic ampoule) should undergo a 100% leak test, and the sealing of other packaging containers should be sampled and inspected according to operating procedures.
Article 78: Product packaging containers sealed under vacuum conditions shall have their vacuum degree checked at an appropriate time determined in advance.
Article 79: External contamination or other defects of sterile drugs shall be inspected one by one. If using the lamp inspection method, the inspection should be carried out under the required conditions, and the continuous lamp inspection time of the lamp inspection personnel should not be too long. The vision of the light inspection personnel should be regularly checked. If other inspection methods are used, this method should be validated, and the performance of the equipment should be regularly checked and recorded.
Conclusion
After reading this article, do you have any understanding of the new GMP cleanroom classification & supervision? Strictly adhering to the above regulations can ensure the safety of sterile drugs. If you have any further questions, Aipak Engineering will provide 24-hour service for you.
Don't forget to share this post!
CONTACT US
Tell us your raw material and project budget to get quotations within 24 hours.
WhatsApp Us: +86 181 7101 8586
Tell us your material or budget, we'll reply you ASAP within 24 hours